Journal of Electrochemical Society Sept. 1976 (page 1294)
Preparation and Applications of Graphite Substrate Lead Dioxide (GSLD)
Anode
K.C. Narasimham and H.V.K. Udupa
Central Electrochemical Research Institute, Karaikudi 623 006, India.
ABSTRACT
GSLD anodes have been developed for obtaining suitable size
anodes for use in high amperage cells in the production of chlorates
and perchlorates. The performance characteristics of the anodes in
the preparation of chlorates and perchlorates are described. The
use of the GSLD anode in other inorganic preparations like bromates
, iodates and periodates is also included.
In recent years the quest for the development of indestructible anodes
either as a substitute for costlier anodes or to increase the life of anodes in electrochemical processes has intensified. Increasing interest in the scientific development of inert and insoluble anodes provided a healthy atmosphere meriting considerable research effort both in the improvement of existing anodes and in the development of new anodes. The complex nature of the evaluation problem stems from the number of variables involved, such as electrode life, operating conditions of the cell, and replacement costs.
Graphite and platinum are widely well-known anodes in electrochemical processes and, less frequently, materials like magnetite, lead, and lead-silver or antimony-silver alloy are employed. But the recent researches on the indestructible or inert anodes are largely centered around the development of:
(a) platinum or its alloy coated over titanium and
(b) coatings of oxides of certain metals on suitable substrates.
The main requirements for an oxide anode are:
(a) the possibility of forming ions of different valences to provide for high electrical conductivity
(b) a high anodic potential at evolution of oxygen
(c) absence of rectifying contacts at the boundary of oxide-metal current lead, and
(d) chemically inert.
The high cost of platinum has prompted several attempts to replace this metal by cheaper material. In the last two decades interest in the use of lead dioxide as anode in the place of platinum for the preparation of inorganic and organic electrochemicals(1) has been very much in evidence as seen by the considerable amount of work carried out to obtain lead dioxide deposits in a form suitable for anodes in the production of chlorates and perchlorates.
The successful development of a suitable GSLD anode for commercial scale operations in chlorates and perchlorates has been engaging the attention of the Central Electrochemical Research Institute. This paper reviews the developmental work on this project. Earlier attempts to prepare the lead dioxide electrodes as well as the difficulties encountered by previous workers have been reviewed earlier by Narasimham and Udupa(2-6). Recently , Carr and Hampson(5) reviewed the studies on the electrodeposition of lead laying emphasis on the kinetics of electrodeposition.
Preparation of GSLD Anode
A survey of the literature shows a continuous interest in the preparation of lead dioxide anodes beginning in 1934. Lead dioxide satisfies the major requirements given above for the oxide anode. Angel and Mellquist(6) reported the deposition of lead dioxide from lead tartrate bath. Though electrolysis of almost all soluble salts of lead(5, 7-10) gives lead dioxide deposit under suitable conditions on the anode, the lead nitrate bath is preferred since it may be readily controlled over long plating periods and also due to the high quality of deposit obtained from the bath over a wide range of operating conditions. Beta_PbO2, which has an oxygen overpotential higher than alpha_PbO2, is obtained from the nitrate bath. Japanese workers(11-16) used this nitrate bath extensively for depositing lead dioxide on nickel or mild steel substrate. When lead nitrate alone is used , a dendrite form of lead is also deposited on the cathodes resulting in a shorting of the electrodes. Even though a diaphragm cell has been suggested(17) to prevent lead deposition on the cathode, the addition of copper salt to the extent of 2 - 3% to the bath for preventing the deposition of lead is by far the most important contribution(12) in the field of electrodeposition of lead dioxide. The copper, being more electronegative than lead in the electronegative series, deposits preferentially on the cathode. Grigger et al.(18)and Schumacher et al.(19) reported the formation of lead dioxide from the nitrate bath on tantalum or platinum-clad tantalum.
Although much work had been done as described above, the preparation of lead dioxide involved certain disadvantages. In the process developed by the Japanese workers(11-16), massive lead dioxide electrodes were prepared by depositing lead dioxide up to 1 CM thick or more, then removing the same from the substrate, and finally cutting the deposit into suitable shapes with either an alundum or carborundum grinding stone. This operation needs special care as the deposit is brittle as well as very hard. The conventional clamp current-contacts produces local heating and hence suitable modifications have to be made.
The successful electrodeposition of lead dioxide from lead-copper nitrate bath on graphite substrate carried out simultaneously by the authors(2, 20, 21) and by Gibson((22, 23) in the U.S. obviates the difficulties experienced by earlier workers. While Gibson used a nonionic surface active agent in the bath, Narasimham and Udupa employed rotation for the cylindrical rods and to-and-fro motion for the plates during deposition to inhibit gas bubbles from sticking to the surface, thereby avoiding pinholes and pitting in the coating. Although hydrodynamic factors, such as the decrease in the thickness of the diffusion layer and also the easy transport of ions to the interphase, do exist during the movement of the electrode, the important aspect in this case happens to be the dislodging of gas bubbles adhering to the surface of the electrode. Similarly, the use of surfactant lowers the interfacial tension, thereby enabling the easy release of gas bubbles from the anode surface. The electrodes thus developed have the following advantages:
a) a thin coating of PbO2 is sufficient(3.5MM)
b) the graphite provides mechanical strength and is completely protected against anodic attack
c) electrical contact can conveniently be made to anode
d) preparation of such anodes for large scale production does not present undue difficulties.
Cell assembly for oxide deposition differed with the size and shape of the anodes required to be coated. The electrolyte contained 325-350 g/L lead nitrate and 25-30 g/L copper nitrate, with an initial pH between 4 & 4.5 . During electrolysis the pH became acidic due to the production of nitric acid and was maintained at 1-1.5 by adjusting the flow rate of the electrolyte. The acid produced was neutralised outside the cell by the addition of lead carbonate or lead monoxide and copper carbonate. Deposition was carried out at current densities of 3-5 A/dM^2 and at a temperature of 58-65C. It is most important to have precleaning operations for the graphite anode prior to deposition, which consist in electrolyzing a 10% (W/V) sodium hydroxide solution with the graphite as anode for 30 min, dipping the anode in 10% (V/V) nitric acid for 10 min , and finally washing it thoroughly with distilled water.
Different rods and plates of GSLD were prepared with different thickness
of deposited PbO2 the thickness of the deposit depending on the process in
which the GSLD is to be used. 1.5 to 2.5mm for chlorate production while
the thickness must be more than 3.5mm for the production of perchlorates.
The addition of different surface active agents to avoid pinholes has been described previously
(13, 14, 25-27). In the course of the study on measurements of stress in electrodeposited lead dioxide(28), the authors found the addition of quaternary ammonium surfactant not only lowered the stress but also helped in obtaining a pore-free deposit on stationary graphite even at higher current densities(29) .
An example of q.a.surfactant is Cetyl Trimethylammonium bromide also called
Hexadecyltrimethylammonium Bromide. Use 2-3 grams per liter.
Applications of GSLD Anodes
Sodium chlorate
A 200A cell with GSLD anode and stainless steel(30) or mild steel(31) cathode was operated continuously; the electrolyte being a solution containing 220g/L sodium chloride and 180-240g/L sodium chlorate with 1N HCl to maintain the pH between 6.4 and 6.8. Though this composition is not critical, but on cyclic use, the solution attained this composition after two or three runs. Table II gives the operating conditions of the cell.
The anodes give satisfactory service for 34 months with 50% of the anodes still in good condition. A current efficiency of 70-80% was obtained while depleting the chloride concentration of electrolyte from 250 to 100g/l. The energy consumption varied from 6.0 to 6.4
kWhr (d.c.)/kg of sodium chlorate(30). When the mild steel was used as container-cum-cathode the anode could be used for 12 months(31) at higher current density, thereby showing almost the same
quantity of production per anode.
| Table I. Operating conditions of chlorate cells using GSLD anode |
|---|
| 200A cell | 800A cell |
|---|
| Characteristics | Cell with SS cathode | Cell with mild steel cathode | Cell with SS cathode | Cell with mild steel cathode |
|---|
| Cell Voltage(V) | 3.2 | 3.2-3.3 | 3.4-3.7 | 3.7-4.2 |
| Anode current density(A/dm^2) | 3.4 | 5.2-5.7 | 4.9 | 10.5 |
| Cathode current density(A/dm^2) | ..... | 2.5-3.0 | 1.4 | 1.13 |
| Current concentration(A/liter) | 6.5 | 15.4 | 3.2 | 2.9 |
| Temperature (C) | 37 | 39 | 40-47 | 40-45 |
| pH | 6.6 | 6.4-6.8 | 6.2-6.8 | 6.2-6.8 |
| Duration of working (months) | 24 | 12 | 24 | 13 |
| Current efficiency (%) | 75-80 | 68-73 | 72-78 | 68-75 |
| Energy consumption(kWhr/kg Na chlorate) | 6.0-6.4 | 6.8-7.2 | 7.0-7.6 | 8.5-9.0 |
Based on the above results, scaling up of the cells to 800A was done for the production of sodium chlorate using GSLD (7.5cm diam. X 60cm long) anodes(32). Two cells were operated continuously, one cell with a concrete container and stainless steel cathode and the other with a mild steel container-cum-cathode. The performance of the cells employing the operating conditions given in table II confirmed the results obtained with 200A cells. The chloride content could be brought down to less than 5g/L in the cell with the stainless steel cathode. No perchlorate formation was observed even at such low chloride concentration. The recent trend in preparation of chlorate is in the use of 30% Iridium-Platinum coated Titanium anodes. Considerable improvements in the efficiency of the electrolytic production of sodium chlorate are claimed for a process developed by Krebs(33, 34). Major differences with usual practice lie in the anodes and in the use of higher current densities (30 A/dm^2)and higher temperatures (> 60C) than current procedures. The consumption of activated alloy is reported to be 400-500mg/ton
of chlorate. The advantage claimed with these anodes is the reduction in the power consumption, viz., 4800-5400 kWhr/ton chlorate(34) as against 6000-6500 kWhr/ton in the existing processes. However, at present the preparation of this anode in India is dependent on the import of all the raw materials.
An electrolytic process for the preparation of sodium chlorate liquor of high concentration (630-660g/L Chlorate, with 5-10g/L chloride) suitable for the in situ production of chlorine dioxide for textile or paper industries, was developed by employing a GSLD anode and stainless steel cathode(35). The high concentration of sodium chlorate could be attained by saturating the cell liquor with solid sodium chloride during electrolysis with a current efficiency of 55-65% and an energy consumption of 8.8-11.5 kWhr (d.c.)/Kg of sodium chlorate. However, the advantage of this method is that the cell effluent can be directly used for the production of chlorine dioxide without processing the liquor for getting solid sodium chlorate and this is possible only with the lead dioxide anode (and perhaps Pt-Ir coating the Titanium anode), not with any other conventional anodes.
Potassium chlorate
Potassium chlorate, which is mainly used in the manufacture of matches, was prepared by the electrolytic oxidation of potassium chloride in the 800A cell using GSLD anodes and stainless steel cathode(36). The cell was operated at an anode current density of 5A/dm^2, a temperature of 55-60 C, and a pH of 6.0-7.0. A current efficiency of 82-85% was obtained with the energy consumption of 6.3-6.5 kWhr (d.c)/kg of chlorate. Efficiency in the production of potassium chlorate higher than the production of sodium chlorate may be attributed to
1) The operation of the cell at higher temperature and consequent lowering of voltage by 0.1 to 0.2V and
2) The conversion of less chloride to chlorate in view of solubility factors.
A 5000A cell(37), which can be considered as a prototype commercial cell, was operated for about 150 days with a GSLD anode employing an anode current density of 15-17 A/dm^2 and a temperature of 50 to 60C. The cell voltage was 3.5 to 3.7V. The higher anode current density has the advantage of using a smaller cell with less initial investment in graphite. The cell effluent, being clear and free from suspended impurities, could be processed further without filtration and pure potassium chlorate (<99%) was obtained by recrystallization.
Another process (38) using a GSLD anode for the production of potassium chlorate was developed on a 800A cell (37) which consisted of the oxidation of sodium chloride to sodium chlorate using a mixed electrolyte consisting of sodium chloride, sodium chlorate,and potassium chlorate in the cell. Potassium chlorate was precipitated by adding solid potassium chloride. The chloride and chlorate concentrations in the electrolyte were maintained between 5 and 1M/liter, respectively, in the feed electrolyte and vice versa in the effluent. The advantage of this method is that a higher concentration of sodium chlorate in liquor can be obtained in the cell which can be reacted with solid potassium chloride to precipitate the potassium chlorate, and the solution, after filtering off potassium chlorate, can be directly used as feed liquor. The novelty of this process is in the working out of the suitable range of compositions of feed and effluent liquor.
Advantages of GSLD anodes in chlorate production:
It has been experimentally proved that GSLD anodes gave the advantage of a life longer than the treated graphite anodes in the chlorate production (consumption of treated graphite is taken as 25-30Kg/ton of chlorate). Small dimensional change of the graphite anodes by the coating of PbO2 does not call for much alteration in the design of the existing cells in industry. With GSLD anodes the cell effluent is clear and hence processing can be carried out without filtration. It is also possible to employ a higher current density which in turn helps in using a smaller cell with less initial investment in graphite.
Perchlorates
Oxidation of sodium chlorate to sodium perchlorate
Due to increasing demand for ammonium perchlorate in rocketry, attention was given to the replacement of costly platinum with GSLD anode in the production of perchlorate. At present, as metal oxide-coated titanium electrodes (like DSA) are not suitable, GSLD anodes are the only alternative to platinum for the production of perchlorate. On the basis of results obtained on laboratory scale (39, 40), two cells of 75A and one of 400A were operated for the production of perchlorate usig the GSLD anode (7.5cm dia. X 30cm long with a deposit of 5mm thick) (41). Six anodes were used in the 400A cell and only one anode in the 75A cell. A saturated solution of sodium chlorate containing NaF (2g/Liter), added to increase the oxygen overvoltage of the anode (13) and also to increase the current efficiency of the process, was electrolyzed using an anode current density of 15 to 25A/dm^2, temperature of 40 to 45C, and pH between 6 and 7. The current efficiency was 70 to 75% with an energy consumption of 3 to 3.3kWhr(dc)/kg of sodium perchlorate. The presence of even small quantities of chromate in the electrolyte affected the current efficiency of the formation of perchlorate at lead dioxide (current efficiency of only 42%), thereby confirming the results of Sugino and Yamashita (42) and Schumacher et al (19). It was pointed out that an insoluble film of lead chromate might be forming on the surface of lead dioxide and thereby hindering the formation of perchlorate. It was found that the thickness of the deposit has a definite effect on the life of the anode and experiments showed that a thickness of 3.5 to 5mm was needed in the preparation of perchlorate.
Direct oxidation of sodium chloride to sodium perchlorate
The methods so far in vogue for the electrolytic preparation of perchlorates involve two steps;
the first stage being the oxidation of chloride to chlorate using graphite (44) or magnetite (45) or lead dioxide anodes (30-32), and the second stage being the oxidation of chlorate to perchlorate using Platinum (46-49) or lead dioxide (18, 19, 39, 40). In between these two stages , the electrolyte has to be processed to isolate the sodium chlorate and recover the unconverted sodium chloride.
Sugino(11) reported the direct oxidation of of chloride to perchlorate using a lead dioxide anode having two discrete electrochemical stages wherein the temperatures of the electrolysis were different and the addition of sodium fluoride was made after all the chloride was converted to chlorate. Udupa et al (50, 52) found that GSLD anode could be used for the preparing of perchlorate directly from sodium chloride in one cell without recourse to processing in between to isolate chlorate.
Large scale trials have been carried out (52) with two cells of 75A and 400A capacity.
The cells were run continuously at an anode current density of 20A/dm^2 and a temperature of
45 to 50C. Six GSLD rods (7.5cm dia. X 30cm long with 4 to 5mm thick coating of PbO2) were used as anodes in the 400A cell and one rod was used in the 75A cell. The electrolyte was a saturated solution of sodium chloride containing NaF (2g/L). The loss due to evaporation was made up with
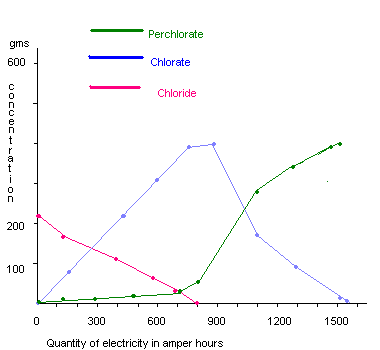 further additions of sodium chloride solution in such a way that the final cell liquor concentration would be 650-700g/liter sodium perchlorate. The graph shows the variation of the concentrations of chloride, chlorate, and perchlorate with duration of the electrolysis. The GSLD anode could be used for more than 450 days of continuous electrolysis in this process. A typical prototype commercial cell of 5000A was designed and operated continuously for 9 months at the optimum conditions (24). A current efficiency of 55-62% with energy consumption of 12.8 to 14.5kWhr/(d.c.)Kg sodium perchlorate was obtained.
further additions of sodium chloride solution in such a way that the final cell liquor concentration would be 650-700g/liter sodium perchlorate. The graph shows the variation of the concentrations of chloride, chlorate, and perchlorate with duration of the electrolysis. The GSLD anode could be used for more than 450 days of continuous electrolysis in this process. A typical prototype commercial cell of 5000A was designed and operated continuously for 9 months at the optimum conditions (24). A current efficiency of 55-62% with energy consumption of 12.8 to 14.5kWhr/(d.c.)Kg sodium perchlorate was obtained.
The novelty of this process is the oxidation of chloride to perchlorate in the same cell without alteration of operating conditions or recourse to intermediate processing. In contemplating the direct oxidation of sodium chloride to sodium perchlorate, none of the commonly used anodes, (graphite, magnetite, or platinum) can be used as a single anode material and GSLD is the natural and economic choice at present. The addition of sodium fluoride can be made either at the beginning of the process or after the conversion of chloride(52).
Sodium perchlorate liquor obtained by both the above methods and having less than 10g/liter sodium chlorate was used for the double decomposition with either potassium chloride or ammonium chloride to give potassium perchlorate or ammonium perchlorate, respectively, and both products conformed to the required specifications (24).
Bromates, iodates and periodates
Based on the resultant conditions of laboratory experiments (53), one 75A cell was run with a GSLD anode for the production of bromates employing an anode current density of 16-20A/dm^2 and a temperature of 55-60C. A current efficiency of 90-95% was obtained with an energy consumption of 4.0-4.5kWhr/(d.c.)Kg sodium bromate. By treating highly soluble sodium bromate liquor with potassium bromide, potassium bromate could be made.
The electrochemical preparation of sodium iodate from iodine was studied using a GSLD anode and a nylon cloth-wrapped stainless steel cathode (54). Based on laboratory scale experiments, one 250A cell was run and a current efficiency of 73-77% was obtained corresponding to an energy consumption of 3.0-3.4kWhr (d.c.)/Kg sodium iodate.
In the oxidation of iodic acid to periodic acid (55), a GSLD anode with rough surface or prepolarized surface of lead dioxide gave better efficiency.
Conclusions
Electrodeposited lead dioxide shows the promise of developing a high efficiency as an inert and insoluble anode in electrochemical processes and, at present, it is the only alternative anode to platinum for the production of perchlorates. Since the current-carrying capacity of GSLD is critical in view of the difference in the coefficients of graphite and lead dioxide, the proper choice of graphite plate or rod has to be made for depositing lead dioxide depending on its subsequent use. The deposition of lead dioxide on other substrates like titanium will have a better future.
REFERENCES
1). H.V.K. Udupa and K.C. Narasimham, J. Indian Chem. Engr., 2, 66 (1960)
2). K.C. Narasimham and H.V.K. Udupa, Proceedings of the Symposium on Electrolytic Cells, Central Electrochemical Research Institute, p 22, Karaikudik, India (1961)
3). K. C. Narasimham, Trans. SAEST, 1, 24 (1966)
4). K. C. Narasimham and H. V. K. Udupa, Chem. Ind. Dev. (Chem. Process. Eng., Bombay), 6, 26 (1972)
5). J. P. Carr and N. A. Hanpson, Chem. Rev., 72, 679 (1972)
6). G. Angel and H. Mellquist, Z. Electrochem., 40, 702 (1934)
7). M. Fleischmann and M. Liler, Trans. Farsday Soc., 54, 1370 (1958)
8). A. N. Kappanna and A. S. Dewagan, J. Indian Chem. Soc., 34, 439 (1957)
9). U. B. Thomas, Trans. Electrochem. Soc., 94, 42 (1948)
10). S. Kiyohara and Y. Shibaski, Japan Pat. 13,370 (1969)
11). K. Sugino, Bull. Chem. Soc. Japan, 23,115 (1950)
12). K. Sugino and Y. Shibazaki, J. Electorchem. Assoc. Japan, 16, 9 (1948)
13). Y. Kato and K. Koizumi, ibid., 2, 309 (1934)
14). Y. Kato, K. Sugino, K. Koizume, and S. Kitahara, Electrotech. J. Japan, 5, 45 (1941)
15). S. Kitahara and T. Osuga, J. Electrochem. Assoc. Japan, 10, 409 (1942)
16). K. Sugino and Y. Shibazaki, Japan Pat. 176,954 (1948)
17). Chemische Fabrik Griesheim-Electron, German Pat.124,512 (1900) and 133,379 (1901)
18). J. C. Grigger, H. C. Miller, and F. D. Loomis, This journal, 105, 100 (1958)
19). J. C. Schumacher, D. R. Stern, and P. R. Graham ibid., 105, 151 (1958)
20). H. V. K. Udupaa and K. C. Narasimham, Indian Pat. 66,195 (1958)
21). K. C. Narasimham, S. Sundararagan, and H. V. K Ududa, J. Electrochem. Soc. Japan, 29, 137 (1961)
22). F. D. Gibson, Jr., U.S. Pat. 2,945,791 (1960)
23). F. D. Gibson, Jr., Chem. Eng., 72, 82 (1965)
24). H. V. K. Udupa, K. C. Narasimham, M. Nagalingam, N. Thiagarajan, R. Palanisamy, S. Pushpavanam, M. Sadagopalan, V. Rangarajand P. D. Jose. ans P. U. John. To be published
25). Y.Shibazaki, J. Chem. Soc. Japan, 57, 794 (1954)
26). J. C. Grigger, U.S. Pat. 2,945, 790 (1960)
27). K. C. Narasimham ahd A. Narayanaswamy, Proceedings of the Symposium on Electrodeposition ad Metal Finishing, India Section of The Electrochemical Society, p118 (1960)
28). K. S. A. Granasekaan, K. C. Narasimham, and H. V.K. Udupa, Electrochem Acta, 15, 1615 (1970)
29). H.V. K Udupa, K. C. Narasimhan and K. S. A. Granasekaran, Indian Pat. 124,215 (1970)
30). H. V. K. Udupa, S. Sampath, K. C. Narasimhan , S. Sundararajan, M. Nagalingam, N. Thiagarajan, P. Govinda Rao, C. J. Jaju, T. J. V. Chanaran, S. Kandasami, G, Subramanian, P. N. N. Nambookri and S. Natarajan, Indian J. Tech., 4, 305 (1966)
31). H. V. K. Udupa, S. Natalingam and others. Chem. Age India, 22, 21 (1971)
32). H. V. K Udups, and others. Indian Journal Tech., 9, 257 (1971)
33). Anon., Process in Eurape, p. 11 (1971)
34) Anon., Platinum Metals Rev., 13, 103 (1969)
35). N. Thaiagarajan, K. C. Narasimham, and H. V. K Udupa, Chem. Ing. Tech., 43, 216 (1971)
36). H. V. K. Udupa and others. J. Applied Chem. Biotech., 24, 47 (1974)
37). K. C. Narasimham, and others. To be published.
38). H. V. K. Udupa and others. Indian Pat. 119,189
39). K. C. Narasimham, S. Sundararajan, and H. V. K. Udupa, This Journal (JES) 108, 798 (1961)
40). K. C. Narasimham and others. Bull. Nat. Inst. Sci. India, 29, 279 (1961)
41). K. C. Narasimham and others. Unpublished data.
42). K. Sugino and M. Yamashita, J. Electrochem. Soc. Japan, 15, 61 (1947)
43). H. V. K. Udupa and others. Indian Pat. 123,290 (1969)
44). P. H. Groggins, A. L. Pitman, J. McLare and F. H. Davis, Chem. Met. Eng., 44, 302 (1937)
45). B.I.O.S. Final report No. 1200.
46). J. C. Schumacher, Trans. Electrochem. Soc., 92, 45 (1947)
47). C. A. Hampel and P. W. Leppla, ibid., 92, 55 (1947)
48). K. C. Narasimham, A. Narayanaswami, and B. B. Dey., J. Sci. Ind. Res. (India) 16A, 512 (1957)
49). H. V. K. Udupa and others. Indian Chem. Eng., 9, 28 (1967)
50). H. V. K. Udupa and others. Indian Pat. 103,067 (1965); U.S. Pat. 3,493,478 (1970)
51). M. Nagalingam and others. Chem. Ing. Tech., 41, 1301 (1969)
52). H. V. K. Udupa and others. J. Applied Electrochem., 1, 207 (1971)
53). S. Sundararajan and others. Chem. Process Eng., 43, 438 (1962)
54). M. S. Venkatachalapathy and others. Electrochem. Technol., 5, 399 (1967)
55). R. Ramaswamy, M. S. Venkatachalapathy, and H. V. K. Udupa, Indian J. Tech., 1, 115 (1963).
**********************************************
HIT THE BACK BUTTON ON YOUR BROWSER